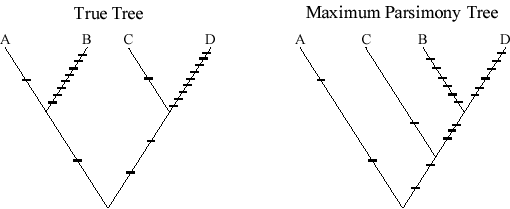
Phylogeny (cont.)
One catch with Maximum Parsimony: If the amount of change along different branches is
quite unequal, such that distant branches both have many more changes than
the intervening branches, then Parsimony is "positively misleading" in
that it will tend to put the long branches together.
Unrooted Trees
If we do not specify the ancestral state of characters we can still construct a tree, but it is unrooted and thus not really a phylogeny.
Example:
Consider these sequences:
S1 = ACTGTT
S2 = GCAGTG
S3 = ATACTG
S4 = ACACAG
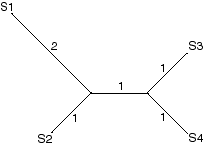
We can construct a diagram illustrating the distance, in terms of number of differences, from each sequence to each other sequence.
S1 and S2 differ by 3 sites.
S1 and S3 differ by 4
S1 and S4 differ by 4
S2 and S3 differ by 3
S2 and S4 differ by 3
S3 and S4 differ by 2
Putting these together produces this tree:
We can convert this into a phylogeny by choosing a root. For example,
if S1 is the outgroup, then the root is between S1 and the node it shares
with S2, producing this rooted tree (with the character state changes mapped on):

Alternately, if we place the root along the central branch, we get:
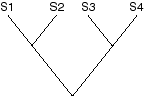
Note that choosing a root essentially decides which character states are ancestral and which are derived.
Homework Problem
1) Draw an unrooted tree for the following sequences:
3) Plot the synapomorphies that unite the different clades onto the tree.
Rooting the large scale tree of life.
Problem: how to root a tree when there is no outgroup (since we are dealing with the tree of all lifeforms on earth)?
Solution: consider a pair of genes that arose from a gene duplication
event that occurred before the common ancestor of all the organisms on our
tree.
Since we know that these genes must have originally been identical,
we can identify derived states (apomorphies) for each.
Example: ATPase-a and ATPase-b appear to have arisen by gene
duplication from a single ancestral gene; however, all extant organisms have
both ATPase-a and ATPase-b.
Catalyze ATP -> ADP
We can draw the phylogeny of these genes within the phylogeny of organisms:
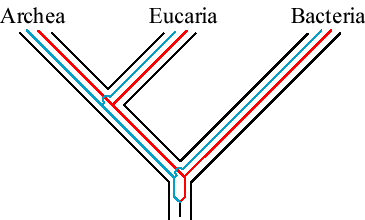
Notice that the topology of the gene tree, taking away the outline of the
species tree, looks like this:
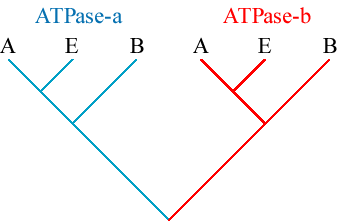
Thus, any modern ATPase-a can be used as an outgroup for all ATPase-b, and vice verse.
Now consider only those parts of the sequences at which ATPase-a and -b are different. These changes must have occurred since the original gene duplication event, and therefore represent apomorphies.
For example, consider these (hypothetical short) sequences:
| ATPase-a (Arch) | TTAGGC |
| ATPase-a (Euk) | TTAGGC |
| ATPase-a (Bac) | CTAGGC |
| ATPase-b (Arch) | CTAGGA |
| ATPase-b (Euk) | CTAGGA |
| ATPase-b (Bac) | CTAGGC |
We can infer that the ancestral state for site 1 is C, and that for site 6 is also C. Since Archea and Eukaryotes share the derived states at these sites, we have evidence that they are closer to one another than either is to Bacteria.
Looking at many sites, we find that Eucaryotes (nuclear genome) and Archaea share far more of these derived changes than either does with bacteria.
The same holds true for other pairs of genes resulting from ancient
duplications.
Thus, we infer that the Archaea and the nuclear genome of Eucaryotes
are more closely related to one another than either is to Bacteria.
Note: We now know of archea (the "Lokiarcheota") that are closer to Eukaryotes than they are to some other Archea.
This means that the Archea, as traditionally defined, are paraphyletic, but it does not change the fact that all Archea are closer to Eukaryotes than any are to the Bacteria
Gene Trees and Species Trees
We generally think of a phylogeny as showing relationships between
species. However, each independently assorting genetic element has its
own history, which may not be the same as that of the species as a whole
or of other genes.
Consider this case:
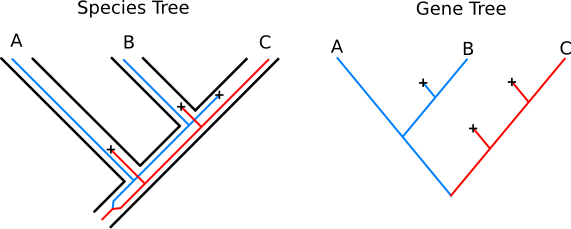
Here, red and blue denote different alleles at the same locus. These alleles persisted in the population through two speciation events, but one or the other went extinct in each of the final species. Note that the history of branching events for this locus has a different shape than the history of branching of reproductively isolated populations.
The gene tree and species tree being different is likely only if:
1) The time between speciation events is small.
Or
2) There has been balancing selection (heterozygote is most fit)
maintaining the polymorphism in the face of drift.
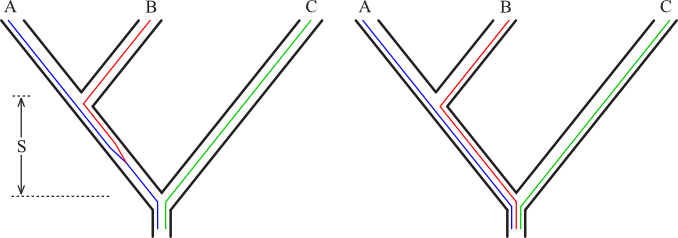
In the case on the left above, the gene tree must match the species tree.
In the case on the right, we could get any of the three possible gene trees (one of which happens to match the species tree) with equal probability.
Thus, if we look at many loci, the gene tree that matches the species tree should appear more often than 1/3 of the time, and the other two trees should appear less than 1/3 of the time and should be equally common.
The time between the speciation events can be estimated from the proportion of gene trees that match the species tree.
If we see many loci at which the gene tree differs from the species tree, then the time between speciation events must have been short (since examples of balancing selection are rare).
This appears to be the case with the Human, Chimp, Gorilla tree.
When we look at hundreds of loci, we get all possible trees, though one is much more common than the other two:

This result strongly supports the hypothesis that humans and chimps are closest relatives.
It also suggests that the time between the branching off of Gorillas and the Human-Chimp split was short; in the range of 0.5-1.5my between the two branching events.
Note: The split between Humans and Chimps was ~6 mya.
Jul 8, 2021