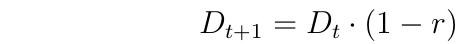
Recall Gametic Disequilibrium (D)
This is a measure of the degree of association between alleles at two different loci.
Thus, if there is any recombination at all, Gametic Disequilibrium declines to zero with random mating and no selection.

Note that r ranges from 0 to 0.5, with r = 0.5 for loci on different chromosomes.
Thus, even for unlinked loci, D does not decline to zero in one generation.
This shows that recombination can drive evolution by changing the frequencies of haplotypes.
Recombination alone does not, however, change the frequencies of alleles.
Genetic Drift
Genetic drift is the change in allele frequencies that occurs in finite
populations due to random variation in which gametes form the next generation.
Drift does not create new alleles, but changes the frequencies of alleles already in the population.
For the moment, we will assume that there is No Selection.
This is the same as saying that we are considering Neutral Alleles
Consider a finite population in which each genotype in the next generation is formed by randomly choosing two alleles that are copies of some alleles in the current generation.
In any given generation, some alleles contribute more copies to the next generation than do others, just due to random sampling.

There are two different, but equally valid, ways of looking at drift: we can look forward in time or backwards in time.
View #1 - Looking forward, Random Walk
One way to think of this process is as a random walk of allele frequency.
Every time an allele contributes more copies to
the next generation than are present
in the current generation,
it takes a step towards fixation.
Every time it contributes fewer copies to the next
generation than are currently present,
it takes a step towards
extinction.
If we take 40 populations, each with 5 individuals (very small) and
each with an initial frequency of the A1 allele of 0.5 (p =
0.5), then plotting the value of p for each population over 40 generations
produces a bunch of curves that look like this:
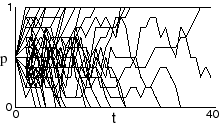
Note that after 40 generations, the allele has gone to fixation or been lost in all but one of the populations.
View #2 - Looking backwards, Gene Lineages
If we just follow the lineages of alleles, we see that the descendant lineages of most loci go extinct, leaving one that ultimately constitutes the entire population.
The most recent common ancestor of all copies of the gene in the population is called the Coalescent point for the entire population. The body of theory that studies it is called "coalescent theory".

Note: For the rest of the discussion, I will use N to denote population size and assume that we are talking about diploid organisms, so there are 2N copies of each locus in the population.
In coalescent theory, we think of a population as a set of gene copies, without regard to how they are arranged into genotypes.
The principal quantity that we work with is the probability that two randomly chosen gene copies coalesce in the previous generation, denoted PC

To find PC, note that the first of our chosen gene copies must have come from some copy in the previous generation. The probability that our two copies coalesce in that generation is just the probability that the second copy comes from that same parent copy. Since there are 2N copies in the parent generation, we get:

The probability that our two copies do not coalesce in the previous generation, PNC, is then just:

The probability that our two copies do not coalesce for n generations (note, I used t in lecture), looking back in time, is then:

Since 1 - 1/(2N) is less than 1, the probability of not coalescing approaches zero as n increases to infinity.
Thus, the probability that our two copies coalesce approaches 1 as we look farther and farther into the past
This Figure shows the results of two computer simulations illustrating coalescence.
Note that such a lineage is for a single, non recombining genetic element, such as a locus or mitochondria. Different such units will have different lineages.
We can view this in two ways:
Using the coalescent approach, we can quickly calculate the probability that a selectively
neutral allele will go to fixation due to drift. The reasoning goes like this:
At any given time, there is a single copy of the gene that will end up
being the common ancestor of all copies at some point in the future. A
particular allele, say A1, will go to fixation if the set of
all A1 alleles includes the lucky gene copy that will be the future
coalescent. Since all gene copies have an equal chance of being the lucky one,
the probability that an A1 allele will be it is just the frequency of
A1 alleles.
P[fixation of a neutral allele with current frequency p] = p
Thus, at any given time, the probability that a particular neutral allele
will go to fixation by drift is just the frequency of that allele at that time.
The frequency of a new mutation is 1/(2N). Thus:
P[fixation of a new neutral mutation by drift] =
1/(2N).
Wish to know: How much evolution is should result from fixation of neutral mutations by drift?
(Rate of fixation) = (rate of appearance) * (prob. of fixation for a new mutant)
Recall that the probability of fixation at any particular time is simply the allele frequency at that time. A new mutant exists as only one copy and thus has a frequency of 1/(2N), which is therefore it's probability of fixation.
Now let un be the per locus neutral mutation rate.
The total rate of appearance of neutral mutants in the entire population is just the per locus rate times the number of loci, or 2Nun .
We thus get:
(Rate of fixation) = 2Nun * 1/(2N) = un .
Thus, the rate of fixation of neutral alleles is just the per locus mutation rate.
This rate tends to be constant across taxa, so we have the basis for a Molecular Clock.
Question: How do we test the constancy of the molecular clock?
Answer: We need a phylogeny and DNA sequence date for the species that we are interested in and one more species, an "outgroup" that is more distantly related to either of our focus species than they are to each other.
We also need the concept of "genetic distance". This is just the number of nucleotide sites at which two species differ.
For example, The following two sequences are separated by a distance of 3 nucleotide
differences (in Blue):
AAGCCAT
and
ACACCGT
Now consider two species, A and B, that differ by 10 mutations. If molecular evolution has been clock like in this group of species, then the rate of molecular evolution should have been the same along the two branches leading from the common ancestor of A and B (labeled "branch 1" and "branch 2" in the figure below). Just given the sequences for A and B, though, we can not determine if this is true.

The key is to consider an outgroup (species O in the figure), another species that split off before the common ancestor of A and B.
In the figure on the left, the total number of changes between O and A is 12 and between O and B is 6 (count the tick marks while tracing the path between two species to get the genetic distance).
This tells us that there has been more molecular evolution along branch 1 than along branch 2.
By contrast, if the distance from O to A is the same as the distance from O to B (as in the figure on the right), then the rate of evolution along branch 1 has been the same as that along branch 2, and the molecular clock is confirmed. Jul 8, 2021